|
CISTI
ARACNOIDEE |
|
Che cosa sono le cisti
aracnoidee?
|
| Consistono di una cavità contenente liquor e rivestita da aracnoide normale e collagene. Sono di origine malformativa. Si sviluppano sulla membrana aracnoidea, una delle tre membrane che rivestono l'encefalo ed il midollo spinale. In 2/3 dei casi si localizzano in regione sopratentoriale, in 1/3 dei casi in regione sottotentoriale; di rado a livello spinale. Sono più frequenti nei maschi. |
|
Le cisti aracnoidee sono frequenti?
|
| Sono assai rare quelle sintomatiche, mentre si possono spesso trovare come reperto occasionale in varie indagini
neuroradiologiche. |
|
Quali sono i sintomi che possono dare?
|
Le cisti aracnoidee si manifestano di solito nell'infanzia e nei giovani prima dei 15 anni di età.
I sintomi sono legati alle dimensioni ed alla localizzazione. Le cisti piccole sono in genere asintomatiche, quelle più voluminose causano sintomi e segni vari. I disturbi più precoci sono in genere cefalea e crisi epilettiche, mentre i deficit focali, come debolezza o paralisi di un lato del corpo o atassia, compaiono più tardi.
Le cisti sottotentoriali, localizzate cioè nella fossa cranica posteriore provocano ostruzione del circolo liquorale con idrocefalo e sintomi di ipertensione endocranica. Nei bambini possono comportare un aumento asimmetrico della circonferenza cranica (macrocefalia), ritardo psicomotorio e turbe comportamentali.
Le cisti localizzate a livello spinale tendono a farsi strada verso la radice, provocando un allargamento del forame di coniugazione. In questi casi possono manifestarsi con una sofferenza monoradicolare, che a livello lombare può simulare un'ernia del disco. |
|
Come sono diagnosticate le cisti
aracnoidee?
|
| La diagnosi si basa sugli esami neuroradiologici (Tomografia Computerizzata e Risonanza Magnetica). La somministrazione di m.d.c. per via intratecale consente di determinare la presenza o meno di una comunicazione della cisterna con lo spazio subaracnoideo. Nei bambini la radiografia del cranio evidenzia l'aumento del volume del cranio e l'assottigliamento della volta cranica a livello della cisti. |
|
Come sono trattate le cisti aracnidee?
|
| Le cisti piccole e asintomatiche non sono trattate. Le cisti voluminose e superficiali sono asportate, mentre quelle a sede profonda sono sottoposte ad un intervento di shunt, che mette in comunicazione la cisti con le normali vie liquorali. Il trattamento chirurgico è seguito dalla regressione dei disturbi clinici. |
|
Qual è l'aspettativa di vita in chi è portatore di cisti
aracnoidee?
|
|
Nei casi non trattati le cisti aracnoidee sintomatiche possono causare un grave danno neurologico permanente dovuto alla progressiva espansione delle cisti o ad emorragia. |
|
CRANIOSTENOSI |
|
Cosa sono le
craniostenosi?
|
Le craniostenosi sono anomalie congenita caratterizzate da prematura saldatura, prima del completamento della crescita encefalica di una o più suture craniche, cioè delle parti che uniscono le ossa del cranio. Questo determina un mancato accrescimento delle regioni corrispondenti della volta cranica, con crescita compensatoria di altre regioni e pertanto deformazione del cranio. Anche l'encefalo non può accrescersi per le ridotte dimensioni del cranio, ne consegue una sofferenza encefalico tanto maggiore quanto più precoce è la chiusura delle suture.
La condizione può rappresentare uno degli aspetti di una sindrome o anormalità genetica o può verificarsi spontaneamente. Alcuni casi possono essere associati con disordini quali microcefalia (in cui il cranio è ridotto di volume per sviluppo deficitario dell'encefalo) ed idrocefalo. |
|
Le craniostenosi sono frequenti?
|
| Sono
rare. |
|
Quali sono i sintomi nelle craniostenosi?
|
Il primo segno è una anomala forma della testa. Frequenti sono i segni oculari: aumento della distanza tra i globi oculari, strabismo divergente, diminuzione dell'acutezza visiva e nel 75% dei casi esoftalmo, cioè protrusione dei globi oculari per riduzione di volume delle cavità
orbitarie.
Nel 30% dei casi è presente un ritardo mentale con sviluppo psicomotorio deficitario. Possono talora comparire i segni e sintomi dell'ipertensione endocranica. Possono anche verificarsi crisi epilettiche e cecità. |
|
Come sono diagnosticate le
craniostenosi?
|
| La periodica misurazione della circonferenza cranica è utile per seguire l'accrescimento del cranio. La radiografia e la Tomografia Computerizzata del cranio confermano le anomalie ossee ed evidenziano la parziale o completa saldatura delle suture. |
|
Come sono trattate le
craniostenosi?
|
| Il trattamento chirurgico va effettuato precocemente per diminuire l'ipertensione endocranica, permettere al cranio di accrescersi unitamente all'encefalo. Oltre che a prevenire i segni di sofferenza encefalica, l'intervento chirurgico ha uno scopo estetico, migliora l'aspetto della testa. |
|
Qual è l'aspettativa di vita nelle
craniostenosi?
|
|
La prognosi varia in rapporto alla presenza di anormalità associate e se sono coinvolte suture singole o multiple. La prognosi è migliore per quei casi con coinvolgimento di singola sutura e nessuna anormalità associata. |
|
MALFORMAZIONE
DI ARNOLD-CHIARI |
|
Cosa è la malformazione di Arnold-Chiari?
|
Consiste nella dislocazione di alcune strutture encefaliche dalla loro normale posizione. È un difetto congenito, cioè presente fin dalla nascita, o acquisito che interessa il tronco encefalico ed il cervelletto. Può essere associato a molte altre anomalie, incluso mielomeningocele, siringomielia e spina bifida.
Si distinguono due tipi di malformazione. In entrambi i tipi, l'area (chiamata fossa cranica posteriore) in cui il cervelletto ed il tronco encefalico sono localizzati, è più piccola del normale. Questo comporta che il cervelletto ed il tronco encefalico sono spinti in basso attraverso una piccola apertura chiamata foro magno.
Nel tipo I le tonsille cerebellari sono dislocate attraverso il forame magno nella parte superiore del canale cervicale. Nel 20-30% dei casi si ha anche siringomielia e in un quarto dei casi anomalie ossee della base cranica.
Nel tipo II sono dislocati nel canale cervicale il verme e la parte inferiore degli emisferi del cervelletto, il bulbo ed il IV ventricolo. Di solito si associa mielomeningocele.
Il tipo II è più grave del tipo I ed il trattamento è più complicato. |
|
Questa malformazione è frequente?
|
| La malformazione è rara. |
|
Quali sono i sintomi nella malformazione di Arnold-Chiari?
|
I sintomi sono legati alle strutture dell'encefalo e alla parte superiore del midollo spinale compresse.
Nel tipo I i sintomi possono manifestarsi solo all'adolescenza o nell'età adulta. L'età media di presentazione è 41 anni. Si può avere: cefalea, dolore al collo, perdita di equilibrio, fastidio agli arti, disturbi visivi, vertigini e difficoltà alla deglutizione. I sintomi possono peggiore con la tosse o con gli sforzi.
Nel tipo II la maggior parte dei pazienti sono bambini. La sintomatologia, spesso drammatica, frequentemente si manifesta dopo la nascita. I sintomi includono deficit degli ultimi nervi cranici, quali turbe della deglutizione, stridore laringeo (cioè difficoltà respiratorie) da paralisi delle corde vocali, disturbi cerebellari (opistotono, nistagmo, atassia), brevi episodi di apnea da disfunzione del tronco encefalico, dolori nucali, parestesie agli arti. Tardivamente si instaura una sindrome da ipertensione endocranica legata all'idrocefalo per compressione del IV ventricolo e delle vie
liquorali. |
|
Come è diagnosticata questa malformazione?
|
| L'indagine di elezione è la Risonanza Magnetica. La Tomografia Computerizzata mostra le alterazioni ossee spesso associate a carico della cerniera
occipito-cervicale. |
|
Come è trattata la malformazione di Arnold-Chiari?
|
| Il trattamento consiste in una decompressione sottooccipitale, quando presenti i segni di compressione delle strutture della fossa cranica posteriore, o la deviazione liquorale per correggere l'idrocefalo. |
|
Qual è l'aspettativa di vita?
|
|
I risultati sono in genere buoni quando la chirurgia è instaurata precocemente.
Nel tipo II il 68% dei casi mostra una completa o quasi risoluzione dei sintomi, il 12% moderati deficit residui ed il 20% non presenta miglioramenti (in genere i neonati hanno un andamento peggiore dei bambini). |
|
MALFORMAZIONE
DI DANDY-WOLKER |
|
Cosa è la malformazione di
Dandy-Wolker?
|
Consiste in una malformazione congenita della fossa cranica posteriore con mancata apertura dei forami di Luschka e Magendie, che connettono il sistema ventricolare con lo spazio subaracnoideo.
Ne consegue enorme dilatazione (o cisti) del IV ventricolo, responsabile di idrocefalo e della mancata fusione degli abbozzi del cervelletto e della formazione del verme. Il verme cerebellare è assente o ipoplasico e gli emisferi sono piccoli. Il tronco encefalico ed il midollo cervicale sono appiattiti.
La sindrome di Dandy-Walker è spesso associato con altre anomalie strutturali del sistema nervoso centrale, come agenesia del corpo calloso e malformazioni della faccia, arti e cuore. |
|
La malformazione di
Dandy-Wolker è frequente?
|
| È rara (2-4% di tutti i pazienti con idrocefalo). Spesso si associa a malformazioni congenite dell'encefalo o di altri organi. |
|
Quali sono i sintomi nella malformazione di
Dandy-Wolker?
|
La sindrome può apparire drammaticamente o essere totalmente asintomatica.
I sintomi si rendono manifesti alla nascita e nei primi mesi di vita. Sono quelli dell'idrocefalo, con aumento del volume cranico, diastasi delle suture e rallentamento dello sviluppo psico-motorio.
Nei bambini più grandi si possono manifestano segni e sintomi di ipertensione endocranica e/o segni di disfunzione cerebellare come atassia (mancanza di controllo muscolare) e nistagmo. Altri sintomi includono: disfunzione dei nervi cranici, bulging occipitale e anormalità respiratorie. |
|
Come è diagnosticata la malformazione di Dandy-Wolker?
|
| Mediante la Tomografia Computerizzata e Risonanza Magnetica. |
|
Come è trattata la malformazione di Dandy-Wolker?
|
| Il trattamento consiste in uno shunt della dilatazione
ventricolare. |
|
Qual è l'aspettativa di vita in tali malformazioni?
|
|
La prognosi è solo moderatamente favorevole, anche quando l'idrocefalo è trattato precocemente. La presenza infatti di multipli deficit congeniti può incidere sulla sopravvivenza. La prognosi per uno sviluppo intellettivo normale è variabile dipendendo dalla gravità della sindrome e dalle malformazioni associate. |
|
ENCEFALOCELE |
|
Cosa sono gli encefaloceli?
|
Per meningocele cranico si intende una malformazione caratterizzata dalla protrusione di una sacca costituita dalle meningi e contenente liquor attraverso un difetto osseo mediano del capo.
Si parla di encefalocele quando la sacca contiene anche tessuto nervoso.
Le sedi più frequenti sono la regione occipitale e quella frontale. Possono raggiungere dimensioni anche enormi. Si evidenziano alla nascita sulla linea mediano. La cute sovrastante mostra spesso anomalie vascolari. |
|
L'encefalocele è frequente?
|
| Sono assai rari sia il meningocele e ancor più
l'encefalocele. |
|
Quali sono i sintomi negli encefaloceli?
|
Danno una tumefazione ricoperta da cute. Quelli localizzati all'interno delle cavità nasali possono causare ostruzione intermittente delle vie aeree; quelli orbitari esoftalmo.
L'esame neurologico, anche nei casi contenenti tessuto nervoso, è in genere negativo. |
|
Come sono diagnosticati gli encefaloceli?
|
La radiografia del cranio evidenzia il difetto osseo.
La Tomografia Computerizzata e la Risonanza Magnetica consentono la diagnosi. |
|
Come sono trattati gli encefaloceli?
|
| L'intervento con riduzione del contenuto della sacca all'interno del cranio e plastica della breccia va attuato al più presto dopo la nascita. Negli encefaloceli occipitali si può sviluppare dopo un certo tempo dall'intervento chirurgico un idrocefalo che necessita di uno shunt di derivazione. |
|
Qual è l'aspettativa di vita negli encefaloceli?
|
|
Il tutto è condizionato all'eventuale danno neurologico associato. |
|
MIDOLLO
ANCORATO |
|
Cosa è il midollo ancorato?
|
| Nel feto il midollo spinale occupa tutto il canale vertebrale. Nell'adulto il rachide risulta più lungo del midollo spinale che termina a livello dello spazio L1-L2. La parte terminale del midollo spinale (cono midollare) e filum terminale può rimane ancorata al fondo del sacco durale (per spina bifida, cisti sacrali, ecc.) e durante l'accrescimento si stira in senso longitudinale, causando danno neurologico. |
|
Il midollo ancorato è frequente?
|
| Il midollo ancorato è assai raro. |
|
Quali sono i sintomi nel midollo ancorato?
|
| Nell'età infantile la sintomatologia è quella della malformazione congenita associata . Nell'età adulta si hanno dolori a livello sacrale, peri-genitale e degli arti inferiori, poi disturbi sensitivi e motori, disturbi
sfinterici. |
|
Come è diagnosticato il midollo ancorato?
|
| La Risonanza Magnetica mostra chiaramente il difetto. La Tomografia Computerizzata utile per evidenziare le anomalie ossee spesso associate. |
|
Come è trattato il midollo ancorato?
|
| Occorre rendere il midollo disancorato e ben mobile attraverso una sezione del filum terminale ispessito al di sotto del cono, nel punto di attacco durale. Il trattamento è indicato nei pazienti con segni neurologici progressivi. |
|
Qual è l'aspettativa di vita in chi presenta un midollo ancorato?
|
|
L'intervento è seguito dalla scomparsa o miglioramento del quadro clinico. |
|
SIRINGOMIELIA |
|
Cosa si intende per
siringomielia?
|
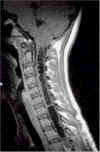
Siringomielia indica la patologica formazione di una o più cavità nel midollo spinale, orientata lungo il suo asse maggiore. Spesso la cavità è situata in prossimità del canale centrale, che a volte è inglobato. La cavità, non rivestita da ependima, interessa, totalmente od in parte la sostanza grigia del midollo spinale. La sede più frequente è a livello cervicale e dorsale. Quando il processo cavitario si estende la bulbo si parla di siringobulbia.
Per idromielia si intende, invece, la dilatazione del canale midollare. L'idromielia è sempre di origine malformativa, mentre la siringomielia può essere congenita (disordine della chiusura del tubo neurale e/o disturbo della dinamica liquorale) o acquisita (complicazione di un trauma, meningite, emorragia, tumore o aracnoidite). In quest'ultimo caso la cavità si sviluppa in un segmento del midollo spinale danneggiato da una di queste condizioni. La forma acquisita è talvolta definita anche siringomielia non comunicante, mentre le cavità congenite possono comunicare o meno con il canale midollare.
La siringomielia è frequentemente associata con altre malformazioni, prima fra tutte la malformazioni di Arnold-Chiari, la spina bifida, il meningocele, il mielomeningocele, le anomalie ossee della cerniera, ecc. |
|
Sono frequenti le
siringomielie?
|
| Sono patologie assai rare. |
|
Quali sono i sintomi nella
siringomielia?
|
Nella forma congenita i sintomi di solito incominciano tra i 25 ed i 50 anni e possono peggiorare con qualsiasi attività che provoca fluttuazioni del liquor, Alcuni pazienti, comunque, possono avere lunghi periodi di stabilità. In alcuni casi si associa un idrocefalo.
Dal momento che il midollo spinale connette l'encefalo ai nervi delle estremità, la siringomielia produce dolore cervicale, diminuzione della forza nei segmenti distali degli arti associata a disturbi sensitivi termo-dolorifici (agli arti ed alla parte più alta del torace, la cosiddetta "anestesia sospesa") e disturbi trofici. Ogni paziente presenta però una differente combinazione di sintomi. Nella forma bulbare possono essere interessati vari nervi cranici e si possono realizzare anche disturbi di tipo vegetativo, mentre nella forma lombosacrale, i disturbi motori, trofici e sensitivi interessano gli arti inferiori e le parti basse del tronco e talora possono essere presenti disturbi sfinterici.
Nella forma acquisita i sintomi possono comparire mesi o persino anni dalla lesione iniziale. |
|
Come sono diagnosticate le
siringomielie?
|
| La diagnosi attualmente è assai agevolo grazie all'uso della Risonanza Magnetica. L'esame radiografico del cranio evidenzia le anomalie ossee spesso associate. |
|
Come sono trattate le siringomielie?
|
|
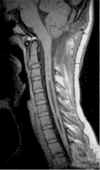
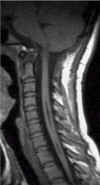
La scelta del trattamento chirurgico dipende dalla causa sottostante. Nei casi con malformazione di Arnold-Chiari si effettua una decompressione occipito-cervicale. Se la causa è un tumore lo si rimuove.
La chirurgia porta alla stabilizzazione o al miglioramento dei sintomi per la maggior parte dei pazienti. Un ritardo nel trattamento può esitare in una lesione irreversibile del midollo spinale.
|
|
|
|
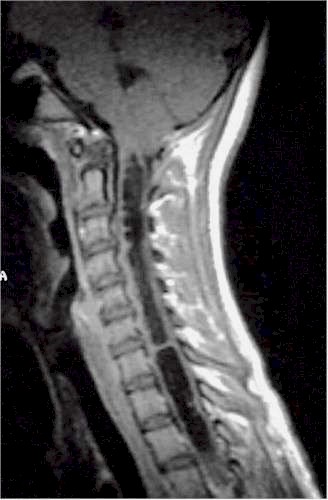
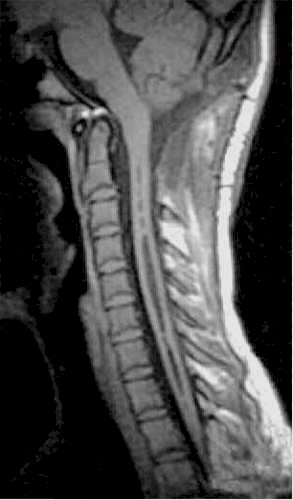
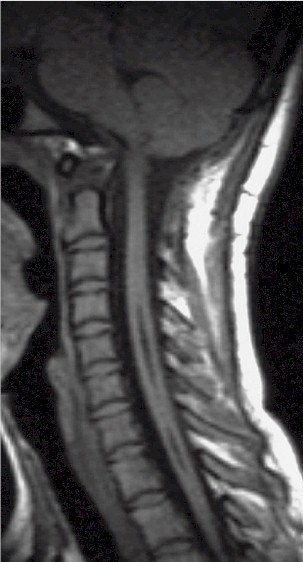
| Siringomielia
cervico-dorsale RM. |
Esito
postchirurgico. |
Due
anni dopo. |
|
|
Nei casi con idrocefalo associato spesso occorre realizzare una derivazione ventricolo-peritoneale.
In assenza di sintomi la siringomielia non è di solito trattata.
|
| Qual è l'aspettativa di vita in chi è affetto da
siringomielia? |
|
Se non trattata chirurgicamente la siringomielia spesso tende ad un peggioramento dei sintomi, perché il decorso è lentissimamente progressivo. In generale la sopravvivenza non è diversa dai soggetti normali, di pari età, ma esiste un alto rischio di invalidità. |
|
SPINA
BIFIDA |
|
Cosa è la spina bifida?
|
Indica un difetto di chiusura, per mancata fusione, della parte mediana di uno o più archi vertebrali con eventuale protrusione di midollo e meningi. Se esiste solo un difetto osseo vertebrale si parla di spina bifida occulta; se si associa una protrusione delle sole meningi meningocele; meningi e tessuto nervoso mielomeningocele. La protrusione del tessuto nervoso impedisce l'ascesa del midollo spinale nel rachide durante la crescita ed il tronco encefalico ed il cervelletto tendono ad erniarsi attraverso il forame occipitale: questo può condurre al decesso o allo sviluppo di idrocefalo.
Le cause sono prevalentemente genetiche. Si localizzano maggiormente a livello lombare e sacrale. |
|
La spina bifida è frequente?
|
| La spina bifida occulta è assai frequente, ha un'incidenza del 10% nella popolazione generale. La sede più frequente è V vertebra lombare e I sacrale. Meningocele e mielomeningocele costituiscono più della metà di tutte le malformazioni del sistema nervoso, con una incidenza compresa tra 0.6-2.5 per 1000 nascite. |
|
Quali sono i sintomi della spina bifida?
|
| La spina bifida occulta, di solito evidenziata accidentalmente durante un esame radiografico del rachide, è di solito asintomatica. Può essere associata ad anomalie cutanee (angioma cutaneo, ciuffo di peli o ad un lipoma). Negli altri casi il quadro neurologico comporta di solito disturbi sfinterici, disturbi motori agli arti inferiori che possono arrivare alla paraparesi o paraplegia, ipoestesia o anestesia con livello superiore lombare o sacrale; frequenti complicazioni infettive dell'apparato urinario ed ulcere da decubito. |
|
Come è
diagnosticata la spina bifida?
|
| La Risonanza Magnetica è l'indagine di elezione. La radiografia del rachide evidenzia la sede e l'estensione del difetto osseo. |
|
Come è trattata la spina bifida?
|
| L'intervento va praticato il più presto possibile, nelle prime 24-48 ore di vita, per prevenire l'aggravamento dei disturbi neurologici ed evitare una infezione meningea se la sacca è ulcerata. La spina bifida occulta non necessita generalmente di trattamento chirurgico. |
|
Qual è l'aspettativa di vita in chi è portatore di spina bifida?
|
| Le alterazioni neurologiche associate possono essere determinanti nel condizionare sia la qualità si la durata della vita. |
|
Le informazioni su patologie e trattamenti neurochirurgici hanno solo scopo informativo.
Non sostituiscono visite o consulti medici.
Se hai problemi di salute, rivolgiti sempre al tuo medico o professionista sanitario di fiducia.
Lo staff di Neurochirurgia è disponibile per chiarimenti, ma non può fornire diagnosi personalizzate
online. |